-
 广州市微生物研究所集团股份有限公司
广州市微生物研究所集团股份有限公司



| 发布时间: | 2024-11-14 |
| 有 效 期: | 9999 |
| 产品规格: | 不限 |
| 所属行业: | 咨询 产品检测服务 |
| 包装说明: | 无 |
| 产品数量: | 9999.00 |
| 价格说明: | 价格:面议 |
 打印本页 打印本页  添加收藏夹 添加收藏夹  点此询价 点此询价
|
|
广州市微生物研究所有限公司以及国内众多的检测公司进行深度合作,共同为国内制造业服务,为中国的制造业开拓海外市场保驾**,为我们日常生活的健康安全树立。
GMP 的主要内容包括哪些方面?
答:可以概括为湿件、硬件、软件。湿件指人员,硬件指厂房、设施与设备,软件指组织、制度、工艺、操作、卫生标准、记录、教育等管理规定。
⑴人员:需有一定数量的技术人员,所有工作人员均需进行知识培训和 GMP 知识培训;
⑵厂房设施要符合 GMP 洁净级别要求,生产药品时必须在洁净区内生产,使用的生产设备要求性与适用性相结合,设备易清洁,不得与药品发生任何变化(一般均采用不锈钢材料制作);
⑶软件:必需制订完善的技术标准、管理标准、工作标准和记录凭证类文件。它包括了生产、技术、质量、设备、物料、验证、销售、厂房、净化系统、行政、卫生、培训等各方面。
GMP(药厂)认证需要洁净检测项目:
1.悬浮粒子
2.浮游菌
3.沉降菌
4.温度
5.相对湿度
6.压差
7.换气次数
8.噪声
9.照度
10.表面微生物
检测标准:
GB/T 16292-2010 医药工业悬浮粒子测试方法
GB/T 16293-2010 医药工业洁净室(区)浮游菌的测试方法
GB/T 16294-2010 医药工业洁净室(区)沉降菌的测试方法
GMP-2010
GB 50457-2019医药工业洁净厂房设计标准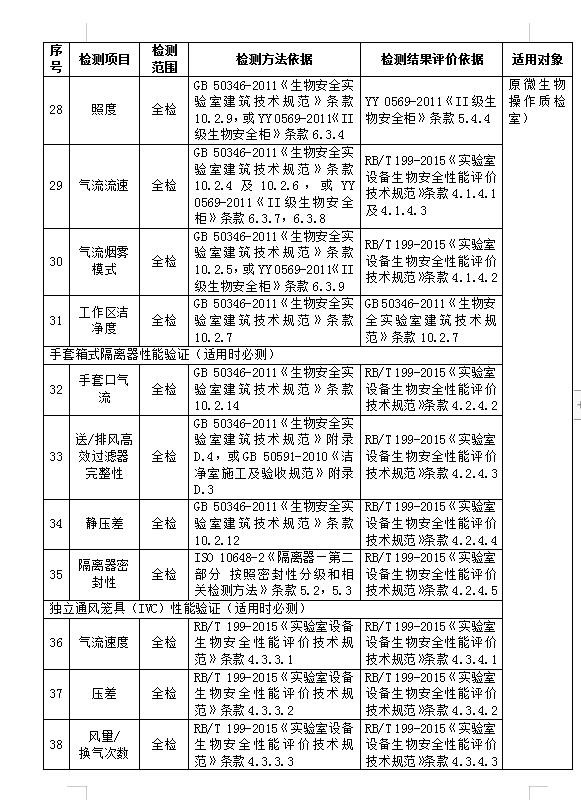
需提交的全部申报材料及数量:
(一)《药品GMP认证申请书》(一式两份)及申请书电子文档。
(二)并附以下相关材料(1份)
1、《药品生产许可证》和营业执照复印件;
2、药品生产管理和质量管理自查情况(包括企业概况及历史沿革情况、生产和质量管理情况,证书期满重新认证企业软、硬件条件的变化情况,前次认证不合格项目的改正情况);
3、企业组织机构图(各部门名称、相互关系、部门负责人);
4、企业负责人、部门负责人简历;依法经过认定的及相关技术人员、工程技术人员、技术工人登记表, 并标明所在部门及岗位;高、中、初级技术人员占全体员工的比例情况表;
5、企业生产范围全部剂型和品种表;申请认证范围剂型和品种表(常年生产品种),包括依据标准、药品批准文号;新药证书及生产批件等有关文件材料的复印件;常年生产品种的质量标准;
6、企业总平面布置图,以及企业周围环境图;仓储平面布置图、质量检验场所平面布置图(含动物室);
7、生产车间概况(包括所在建筑物每层用途和车间的平面布局、建筑面积、洁净区、空气净化系统等情况。其中对β-内酰胺类、避孕药、类、抗类、等的生产区域、空气净化系统及设备情况进行重点描述),设备安装平面布置图(包括更衣室、盥洗间、和物流通道、气闸等,并标明人、物流向和空气洁净度等级);空气净化系统的送风、回风、排风平面布置图;
8、认证剂型或品种的工艺流程图,并主要过程控制点及控制项目;
9、关键工序、主要设备、制水系统及空气净化系统的验证情况;
10检验仪器、仪表、量具、衡器校验情况;
11、企业生产管理、质量管理文件目录;
12、企业符合消防和环保要求的文件;
13、药品委托检验协议及前处理提取委托加工批件的复印件;
14、新开办药品生产企业、药品生产企业新增生产范围申请药品GMP认证,除报送以上资料外,还须报送认证范围涉及品种的批生产记录复印件;
申请企业应当对其申报材料全部内容的真实性负责。以上材料统一用A4纸打印装订成册。
GMP认证需要做什么?
1) 申报企业提交申报资料;
2) 省局对申报材料进行形式审查;
3) 省局对试验及有关原始资料进行现场核查(5个工作日);
4) 国家局审评中心对申报资料进行审评;
5) 企业提出现场检查申请(6个月内)
6) 认证中心组织实施认证现场检查(30个工作日);
7) 现场检查报告报国家局审评中心(10个工作日);
http://gzjczx.cn.b2b168.com
